 |
Marcin Nowotny, PhD, Professor
Correspondence address:
Laboratory of Protein Structure
International Institute of Molecular and Cell Biology
4 Ks. Trojdena Street, 02-109 Warsaw, Poland
Email: This email address is being protected from spambots. You need JavaScript enabled to view it.
tel: +48 (22) 597 0717; fax: +48 (22) 597 0715
|
DEGREES
2020 - Professor of Biological Sciences, nomination by the President of the Republic of Poland
2013 - DSc Habil in Molecular Biology, Institute of Biochemistry and Biophysics, Warsaw, Poland
2002 - PhD in Biochemistry summa cum laude, Nencki Institute of Experimental Biology, Polish Academy of Sciences, Warsaw, Poland (Supervisor: Jacek Kuźnicki)
1998 - MSc in Organic Chemistry and Biochemistry, Department of Chemistry, Warsaw University, Poland
PROFESSIONAL EMPLOYMENT
2008-Present - Professor, Head of the Laboratory of Protein Structure, International Institute of Molecular and Cell Biology in Warsaw, Poland
2017-2019 - Co-founder and Chief Scientific Officer, ProBiostructures, International Institute of Molecular and Cell Biology research service center for pharmaceutical industry
2016-2018 - Deputy Director for Science, International Institute of Molecular and Cell Biology in Warsaw, Poland
POSTDOCTORAL TRAINING
2003-2008 - Postdoctoral Fellow, Wei Yang Laboratory, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, USA
MEMBERSHIP AND AWARDS
2020 - Chair, Scientific Policy Committee, Ministry of Science and Higher Education, Poland
2019 - Member, European Molecular Biology Organization
2019 - Member, Academia Europaea
2018 - Member, Scientific Policy Committee, Ministry of Science and Higher Education, Poland
2018 - MAESTRO, National Science Centre
2016 - TEAM, Foundation for Polish Science
2015 - Jan Karol Parnas Award for the best Polish biochemical publication (with the group of Prof. Janusz M. Bujnicki)
2013 - Academia Europaea Burgen Scholar
2013 - Knight’s Cross of the Order of Polonia Restituta
2012 - Polish Prime Minister Award for scientific achievement
2012 - Ideas for Poland Award, Foundation for Polish Science
2012 - Jan Karol Parnas Award for the best Polish biochemical publication
2012 - International Senior Research Fellowship, Wellcome Trust (renewal)
2012 - Early Career Scientist Award, Howard Hughes Medical Institute
2011 - ERC Starting Grant (2012-2017)
2007 - EMBO Installation Grant
2007 - International Senior Research Fellowship, Wellcome Trust
2003 - Prime Minister Award for PhD thesis
2001, 2002 - START Scholarship for Young Scientists, Foundation for Polish Science
DOCTORATES DEFENDED UNDER LAB LEADER’S SUPERVISION
M. Jaciuk, M. Miętus, M. Czarnocki-Cieciura, M. Śmietański, M. Rażew
1. RNases H
Bacterial RNase H2
RNase H2 cleaves the 5′ phosphate of ribonucleotides in RNA-DNA junctions. It is the only known enzyme that can initiate the process of mutation-free removal of single ribonucleotides from genomic DNA. Such ribonucleotides are very often misincorporated by replicative polymerases and lead to genomic instability.
Rychlik MP, Chon H, Cerritelli SM, Klimek P, Crouch RJ, Nowotny M. Crystal Structures of RNase H2 in complex with nucleic acid reveal the mechanism of RNA-DNA junction recognition and cleavage. Mol. Cell, 2010; 40:658-670
-
First crystal structure of substrate complex of RNase H2 obtained using T. maritima protein.
-
Unique catalytic mechanism in which the substrate is deformed, so that the phosphate group of the RNA-DNA junction participates in metal ion coordination at the active site. This metal ion positions and activates the nucleophilic water molecule. Hence, the substrate is used to assemble the active site to promote its own cleavage.
The studies of RNases H2 were performed in collaboration with Dr. Robert Crouch (National Institutes of Health, USA).

Crystal structure of T. maritima RNase H2 in complex with the nucleic acid substrate. The catalytic domain is in orange and purple and the helical C-terminal extension in yellow. DNA is shown in blue and single ribonucleotide in red. Metal ions at the active site are shown as green spheres.
Human RNase H2
Human RNase H2 comprises three subunits – the catalytic subunit is very similar to monomeric bacterial and archaeal enzymes while the remaining two subunits do not share sequence similarity with any other known proteins. Mutations in human RNase H2 lead to a severe genetic autoimmune disease - Aicardi-Goutières syndrome.
Figiel M, Chon H, Cerritelli SM, Cybulska M, Crouch RJ, Nowotny M. The structural and biochemical characterization of human RNase H2 complex reveals the molecular basis for substrate recognition and Aicardi-Goutieres syndrome defects. J. Biol. Chem., 2011; 286:10540-50
 Crystal structure of human RNase H2. The catalytic subunit is shown in yellow and the auxiliary ones in purple in blue.
Crystal structure of human RNase H2. The catalytic subunit is shown in yellow and the auxiliary ones in purple in blue.
RNase H3
RNases H3 are present in certain bacterial and archaeal species. They share high sequence and structure similarity with RNases H2, however in terms of biochemical properties they are similar to RNases H1 – they prefer to cleave RNA/DNA hybrids in the middle of the RNA sequence. RNase H3 contains a unique N-terminal domain related to TATA-binding protein.
Figiel M, Nowotny M. Crystal structure of RNase H3-substrate complex reveals parallel evolution of RNA/DNA hybrid recognition. Nucleic Acids Res., 2014; 42(14):9285-94
-
The first crystal structure of RNase H3 in complex with the RNA/DNA substrate.
-
The RNA strand is recognized by contacts between 2’-OH groups of four consecutive ribonucleotides.
-
The DNA strand is recognized by deformation to B-form sugar puckers only allowed for DNA.
-
The N-domain specifically binds RNA/DNA hybrid by recognizing 2’-OH of RNA and forming stacking interactions with the ribose rings of the DNA.
-
The mechanism of N-domain is very similar to the structurally unrelated hybrid-binding domain present in the N-terminus of RNases H1. This is a likely case of convergent evolution of RNA/DNA recognition.

Crystal structure of RNase H3 (catalytic domain in yellow and N-domain in green) interacting with an RNA/DNA hybrid (RNA in red and DNA in blue). The cleaved phosphate is shown as a red sphere and the phosphate group of the deformed DNA residue as a blue sphere.
2. DNA repair
Bacterial Nucleotide Excision Repair
Nucleotide excision repair (NER) is one of the major pathways of DNA repair and its main feature is its ability to recognize a wide spectrum of DNA lesions of various sizes and structures. In bacterial NER UvrA, a dimeric ATPase, plays the role of a DNA damage sensor. Damage verification is performed by UvrB helicase and UvrC double nuclease excises the damaged DNA fragment.
Jaciuk M, Nowak E, Skowronek K, Tanska A, Nowotny M. Structure of UvrA nucleotide excision repair protein in complex with modified DNA. Nature Struct. Mol. Biol., 2011; 18:191-197
-
The first crystal structure of UvrA interacting with damaged dsDNA obtained using the protein from T. maritima.
-
UvrA does not interact with the damage site directly but senses the deformed conformation of the DNA induced by the presence of the lesion – bending, stretching and unwinding.

Crystal structure of UvrA dimer (one monomer in color, the other in gray). One ATPase module is shown in cyan and red and the other in pink and blue. The DNA-binding domain is in green and UvrB-binding domain in yellow. Structural zinc ions are shown as orange spheres.
Jaciuk M*, Swuec P*, Gaur V*, Kasprzak JM, Renault L, Dobrychłop M, Nirwal S, Bujnicki JM& , Costa A&, Nowotny M&. A combined structural and biochemical approach reveals translocation and stalling of UvrB on the DNA lesion as a mechanism of damage verification in bacterial nucleotide excision repair. DNA Repair, 2019; 85, 102746; * - equally contributing, & - corresponding authors
-
Computational structural model of UvrA—UvrB—DNA complex involved in DNA damage verification in bacterial DNA repair was prepared and corroborated experimentally by electron microscopy.
-
UvrB uses a β-hairpin element to clamp one DNA strand, each one of the two UvrB molecules in the complex clamps a different DNA strand.
-
UvrB translocates in 3′ direction toward the DNA lesion where the UvrB molecule clamping the damaged strand stalls and recruits UvrC nuclease.
-
This mechanism explains how the initial imprecise localization of the damage by UvrA is converted to precise and strand-specific localization to promote accurate incisions by UvrC.
The studies of damage verification in bacterial NER have been performed in collaboration with Janusz M. Bujnicki (IIMCB) and Alessandro Costa (The Crick Institute).

Model of damage verification in bacterial NER. (A) Model of UvrA2—UvrB2—DNA complex. UvrA dimer shown in two shades of blue. UvrB shown in orange. DNA shown in black. (B) Proposed mechanism. The UvrA dimer bound at the site of DNA modification recruits two UvrB molecules. Each UvrB molecule clamps a different DNA strand under the B-hairpin element (upper panel). Both UvrB molecules then translocate toward the lesion with 5′ to 3′ polarity on the strand under the hairpin. The UvrB molecule that clamps the modified strand will stall at the lesion (green star indicates the site of DNA modification) and the other UvrB molecule (light orange) will dissociate (middle panel). The stalled UvrB recruits UvrC double nuclease (shown in yellow), which makes two incisions indicated with triangles.
Rad2
Eukaryotic nucleotide excision repair (NER) is one of the major DNA repair pathways. It involves the excision of the DNA fragment containing the damage. This is achieved through the action of two nucleases – XPF-ERCC1 complex and XPG (Rad2 in yeast). Rad2/XPG belongs to flap endonuclease metal ion-dependent enzymes along with FLAP1 and EXO1. Its unique feature within this family is the ability to cleave DNA bubbles – substrates with melted single stranded region flanked with double-stranded stretches of DNA.
Miętus M, Nowak E, Jaciuk M, Kustosz P, Studnicka J, Nowotny M. Crystal structure of the catalytic core of Rad2: insights into the mechanism of substrate binding. Nucleic Acids Res., 2014; 42(16):10762-75.
-
The first crystal structure of the catalytic core of Rad2 using a truncated version of the S. cerevisiae enzyme.
-
The main substrate specificity determinant of Rad2 is the interaction of the last exposed base pair of the double-stranded region with the so-called hydrophobic wedge of the enzyme. No interactions with the single stranded portion of the substrate are observed.
-
The main DNA-binding element is potassium-coordinating helix-two-turn-helix (H2TH) motif. It contains an additional charged helix binding the DNA, which is a unique feature of Rad2.
-
The likely explanation for the unique ability of Rad2 to clave DNA bubbles (substrate without a free 5’ DNA end) is the altered structure of the so-called helical arch. In FEN1 and EXO1 this element blocks the exit from the active site preventing cleavage of substrates without a free 5’ end. In Rad2 the helical arch has a different structure forming an exit route from the active site.
 Crystal structure of Rad2-DNA complex. The complex contains two independent protein molecules – one is shown in color: cyan for H2TH motif, green for hydrophobic wedge and orange for helical arch. The DNA is shown in cyan and blue, potassium ion as a purple sphere and the calcium ion at the active site as a green sphere.
Crystal structure of Rad2-DNA complex. The complex contains two independent protein molecules – one is shown in color: cyan for H2TH motif, green for hydrophobic wedge and orange for helical arch. The DNA is shown in cyan and blue, potassium ion as a purple sphere and the calcium ion at the active site as a green sphere.
RuvC
RuvC is a canonical bacterial Holliday junction (HJ) resolvase, which functions as a dimer. HJ are four-way DNA structures formed by the exchange of strands between two helices. They are intermediates in homologous recombination, a process which is used to repair dangerous DNA lesions such as double-strand breaks.
Gorecka, KM, Komorowska W and Nowotny M. Crystal structure of RuvC resolvase in complex with Holliday junction substrate. Nucleic Acids Res., 2013; 41(21):9945-55
-
The first crystal structure of RuvC in complex with a DNA substrate and the first substrate complex structure of a cellular resolvase, solved at 3.8 Å resolution.
-
HJ in a novel tetrahedral conformation with two phosphate groups symmetrically located 1 nt from the HJ exchange point interacting with two active sites of RuvC dimer.
-
Novel mode of HJ recognition relative to phage enzymes for which complex crystal structures had been available.

Crystal structure of RuvC in complex with Holliday junction. The two protomers are shown in pink and orange. The DNA is in blue with cleaved phosphates indicated with spheres.
Górecka KM*, Krepl M*&, Szlachcic A, Poznański J, Šponer J, Nowotny M&. RuvC uses dynamic probing of the Holliday junction substrate to achieve sequence specificity and efficient resolution. Nature Commun. 2019; 10(1):4102; *- equally contributing,
& - corresponding authors
-
Mechanism of sequence specificity and cut coordination revealed by a combination of structural biology, biochemistry, and a computational approach.
-
Correct positioning of the substrate for cleavage requires conformational changes within the bound DNA, which are possible only for the cognate sequence.
-
The conformational changes and the relieving of protein-induced structural tension of the DNA facilitates coordination between the two cuts.
-
The unique DNA cleavage mechanism of RuvC demonstrates the importance of high-energy conformational states in nucleic acid readout.
Studies of RuvC mechanism have been done in collaboration with Jiří Šponer (Institute of Biophysics, CAS)
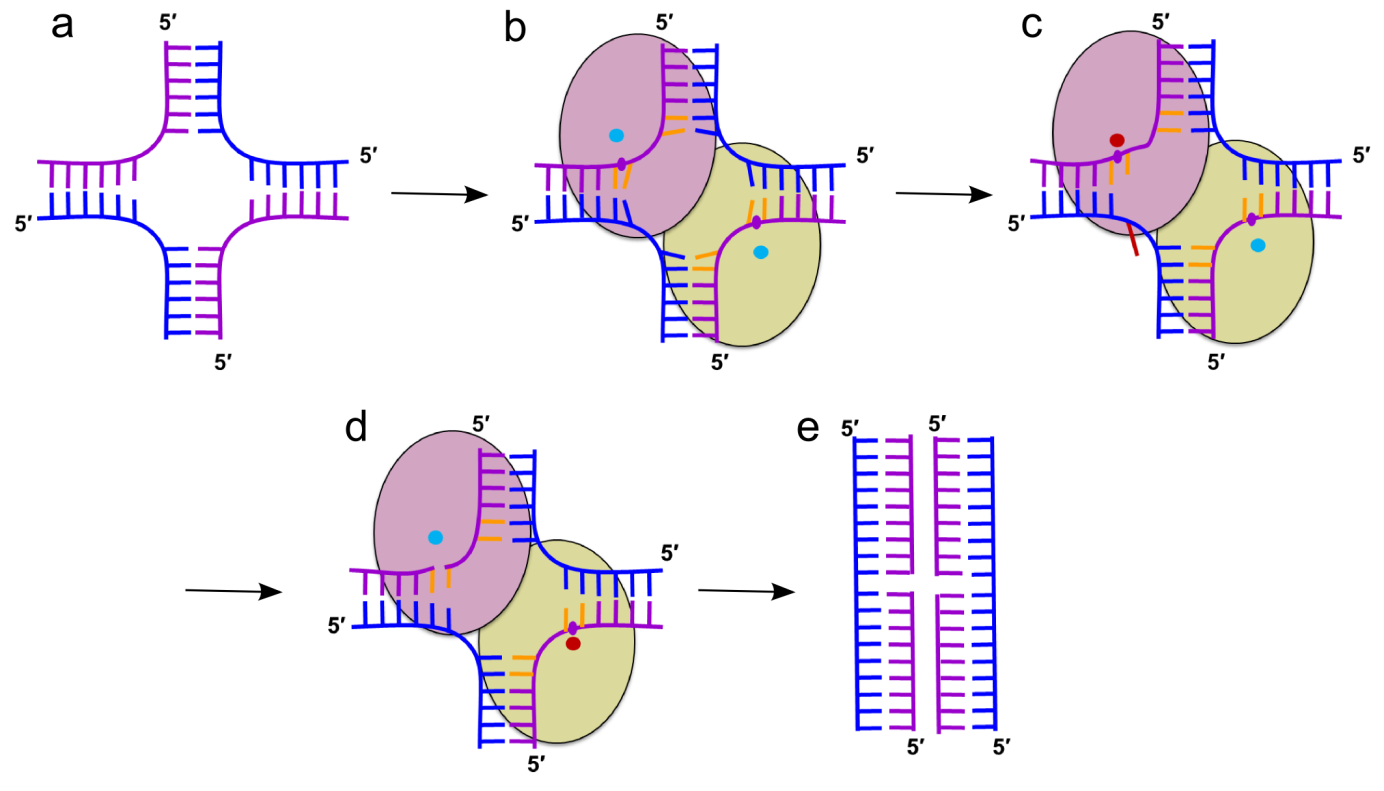
Cartoon representation of the mechanism of HJ resolution by RuvC. (a) Holliday junction. Cleaved and non-cleaved DNA strands are shown in purple and blue ladder-like representations, respectively. (b) Binding of the HJ DNA. The subunits of the dimer are shown as yellow-green and pink ovals. The scissile phosphate is marked as a purple circle. Cyan circles show active sites in an inactive configuration. (c) Flipping of the adenine (red) opposite the scissile base. The active site in the catalytic configuration is shown as a red circle. (d) The second cut. (e) Resolution products
Slx1-Slx4
Slx1 is a nuclease which cleaves various DNA structures during DNA repair and recombination. It associates with Slx4 platform protein which coordinates the action of multiple proteins. Slx1 together with Mus8-Eme1 nuclease constitute one of the major Holliday junction pathways in eukaryotes.
Gaur V, Wyatt HD, Komorowska W, Szczepanowski RH, de Sanctis D, Gorecka KM, West SC, Nowotny M. Structural and Mechanistic Analysis of the Slx1-Slx4 Endonuclease. Cell Rep., 2015; S2211-1247(15)00165-5
-
First structural information for Slx1 and Slx4CCD.
-
Fungal Slx1 forms a homodimer in which the active site is blocked, explaining why Slx1 alone is inactive.
-
Slx4CCD domain binding is mutually exclusive with homodimerization.
-
Slx4 binding exposes the active site of Slx1 and activates the nuclease. This mechanism ensures that the promiscuous and potentially dangerous Slx1 nuclease is only active when bound to Slx4 platform which regulates its activity and coordinates it with other proteins.

Crystal structures of Slx1 homodimer and Slx1 in complex with Slx4CCD domain (orange). Slx1 comprises GIY-IYG nuclease domain (yellow) and RING finger zinc-binding domain (green). Upon Slx4 binding the active site of the nuclease domain is exposed and the enzyme is activated.
Gaur V, Zajko W, Nirwal S, Szlachcic A, Gapińska M, Nowotny M. Recognition and processing of branched DNA substrates by Slx1-Slx4 nuclease. Nucleic Acids Res., 2019; 47:11681-11690
-
Based on a crystal structure, modeling, and biochemical studies, a mechanism was proposed to explain the specificity of Slx1 towards a wide range of branched DNA substrates.
-
Slx1 bends the DNA and identifies the branch point as a flexible discontinuity.
-
Cuts are introduced on the 3ʹ side of the branch point.

Models of Slx1–Slx4CCD3 interactions with branched DNA. (A) Schematic of the DNA flap substrates with terminology of the strands. (B) Model of Slx1–Slx4CCD3 bound to a 3′-flap substrate in configuration, which is conducive to incision in the continuous strand. (Left) Slx1 GIY-YIG domain is shown in yellow with -strands in orange and RING domain shown in blue. Slx4CCD is shown in green. The modeled DNA is based on the structure of R.Eco29kI restrictase (PDB ID: 3NIC) and is shown in black with the scissile phosphate shown as a sphere. A fragment of the DNA that is observed in the Slx1–Slx4CCD-DNA structure is shown in cyan. The possible link between the two DNA double helices is shown as a dashed cyan line. Residues of the active site are shown as red sticks. The same model with protein in surface representation, colored according to the surface potential (±3 Kt/e). (C) Model of Slx1–Slx4CCD3 bound to a 5′-flap substrate, in configuration which is conducive to incision of the flap strand. The representations are the same as in (B).
3. Reverse transcriptases
XMRV reverse transcriptase
Reverse transcriptases use two enzymatic activities – DNA polymerase and RNase H to catalyze the conversion of single-stranded RNA to double-stranded DNA, a process essential for proliferation of retroviruses such as HIV and retrotransposons. Retroviral RTs are divided into two classes – dimeric (i. e. HIV) or monomeric (i. e. gammaretroviral enzyme from mouse Moloney leukemia virus and closely related XMRV).
Nowak E, Potrzebowski W, Konarev PV, Rausch JW, Bona MK, Svergun DI, Bujnicki JM, Le Grice SF, Nowotny M. Structural analysis of monomeric retroviral reverse transcriptase in complex with an RNA/DNA hybrid. Nucleic Acids Res., 2013 Apr 1;41(6):3874-87
-
The first crystal structure of a monomeric RTs in complex with RNA/DNA hybrid visualizing the polymerase-connection fragment of the enzyme.
-
Full-length protein modelled based on small-angle X-ray scattering data.
-
SAXS data demonstrated that the RNase H domain is mobile and only occasionally interacts with the substrate to cleave RNA. This is the mechanism to fine tune RNase H activity.
-
This mechanism of RNase H fine-tuning is different from dimeric RTs which use substrate deformations for that purpose.
 Model of the full length XMRV RT based on SAXS data, a crystal structure of the polymerase-connection fragment in complex with RNA/DNA hybrid (blue-fingers, red-palm, green-thumb, yellow-connection) and the structure of isolated XMRV RNase H domain (orange, Zhou D, J Struct Biol. 2012). RNA template stand is in purple and DNA primer strand in blue.
Model of the full length XMRV RT based on SAXS data, a crystal structure of the polymerase-connection fragment in complex with RNA/DNA hybrid (blue-fingers, red-palm, green-thumb, yellow-connection) and the structure of isolated XMRV RNase H domain (orange, Zhou D, J Struct Biol. 2012). RNA template stand is in purple and DNA primer strand in blue.
Ty3 reverse transcriptase
Retrotransposons are mobile genetic elements that replicate with an RNA intermediate. Reverse activity of element-encoded RT is essential for this process. Retroelements are one of the most potent forces shaping eukaryotic genomes – more than 40% of human genome derives from those elements. Ty3 is a yeast retrolement from long-terminal class termed Ty3/Copia. It is thought that retroviruses evolved from this class of retrotransposons.
Nowak E, Miller JT, Bona MK, Studnicka J, Szczepanowski RH, Jurkowski J, Le Grice SFJ&, Nowotny M&. Ty3 reverse transcriptase complexed with an RNA-DNA hybrid shows structural and functional asymmetry. Nature Struct. Mol. Biol., 2014; 21(4):389-96;
& - corresponding authors
-
The first crystal structure of a retrotransposon RT.
-
Ty3 RT forms an asymmetric homodimer in which one subunit has the polymerase competent configuration and the other has an altered conformation and harbors the RNase H activity.
-
RNase H is postulated to undergo a conformational change to reach the position required for RNA/DNA cleavage, which regulates this activity.
-
Ty3 and HIV RT architecture differs: HIV enzyme is a constitutive heterodimer with both polymerase and RNase H activities residing in the larger subunit and Ty3 is a substrate-induced homodimer in which the two activities are located in different subunits.
-
The studies of XMRV and Ty3 reverse transcriptases have been performed in collaboration with Dr. Stuart Le Grice (National Institutes of Health, USA).

Crystal structure of Ty3 reverse transcriptase. The subunit with polymerase-competent configuration is shown in darker color (blue-fingers, red-palm, green-thumb, yellow-RNase H) and the subunit with altered conformation in lighter shades of the same colors. RNA template stand is in purple and DNA primer strand in blue. Active site residues for polymerase and RNase H domain are shown as sticks.
HIV-1 reverse transriptase
HIV-1 RT is an important drug target in therapy of HIV-1 infection. The RT uses its polymerase and RNase H activities to catalyze the process of reverse transcription, in which the single-stranded RNA of the virus is converted into double-stranded DNA that can be integrated into the host cell genome.
Figiel M, Krepl M, Poznański J, Gołąb A, Šponer J, Nowotny M. Coordination between the polymerase and RNase H activity of HIV-1 reverse transcriptase. Nucleic Acids Res., 2017; 45(6):3341-3352.
-
Dynamics of HIV-1 RT-RNA/DNA complex were studied using a combination of chemical cross-linking and molecular dynamics simulations.
-
The RNA/DNA substrate can simultaneously interact with the polymerase and RNase H active sites.
-
Untwisting of the RNA/DNA substrate double helix is required for its productive interaction with the RNase H active site.
-
This allows HIV-1 RT to regulate the amount of the RNase H activity.

Molecular dynamics simulation of HIV-1 RT RNA/DNA complex (two views). Superposition of the starting model (light colors) and the final model in MD simulations (dark colors). Domains of HIV-1 RT are labeled. RNA and DNA strands of the substrate are shown in red and blue, respectively.
In order to be integrated into the host cell genome, the single-stranded RNA of HIV-1 is converted into double-stranded DNA. The synthesis of the (+)-strand DNA starts from the polypurine tract (PPT) primer. The PPT primer is generated by the RNase H domain of HIV-1 RT which cuts specifically at its termini, but leaves the body of the PPT intact.
Figiel M, Krepl M, Park S, Poznański J, Skowronek K, Gołąb A, Ha T, Šponer J, Nowotny M. Mechanism of polypurine tract primer generation by HIV-1 reverse transcriptase. J Biol. Chem., 2018; 293(1):191-202.
-
Factors involved in recognition of the PPT sequence by HIV-1 RT-RNA/DNA were studied using a combination of chemical cross-linking, molecular dynamics simulations, and single-molecule assays.
-
The PPT is specifically recognized after the complex with HIV-1 RT is formed and not at the stage of binding.
-
Recognition of the PPT is based on two elements: agreement with the sequence preference of RNase H and the indirect readout of the poly-rA/dT stretch. The rigid but brittle poly-rA/dT is not compatible with the catalytically relevant substrate geometry and is prone to undergo sequence slippage upon deformation.
-
The poor match with the RNase H sequence preference and the dynamic properties of the PPT explain the protection of its body from cleavage.
Studies of HIV-1 RT have been done in collaboration with Jiří Šponer (Institute of Biophysics, CAS) and Taekjip Ha (Johns Hopkins Univ.).

Model of PPT recognition by HIV-1 RT. Cleavage at the expected site involves both preferred residues at the cleavage consensus position and the ability of PPT sequence to undergo the appropriate conformational change without distortion (left panel). Cleavage in the middle of the PPT body (A-tract) is inhibited by three elements: non-preferred residues in the consensus positions, rigidity of the poly(rA-dT) sequence, and misalignment of the substrate at the RNAse H active site due to poly(rA-dT) sequence slippage (right panel).
4. RNA processing
Human cap 2’-OH methyltransferase
mRNA contains a cap structure on its 5' terminus. In higher eukaryotes the first and the second ribonucleotide of the body of the mRNA are methylated on the 2' oxygen. This is thought to serve as a tag to distinguish self mRNA from the mRNA of invading viruses. To circumvent the mechanism, some viruses such as West Nile and yellow fever viruses encode their own 2'-OH methyltransferases.
Śmietanski M*, Werner M*, Purta E, Kamińska KH, Stepiński J, Darżynkiewicz E, Nowotny M&, Bujnicki JM&. Structural analysis of human 2’-O-ribose methyltransferases involved in mRNA cap structure formation. Nat. Commun., 2014 Jan 9;5:3004; * - equally contributing, & - corresponding authors
The studies of cap 2'-OH methyltransferase have been performed in collaboration with Prof. Janusz M.Bujnicki (IIMCB).
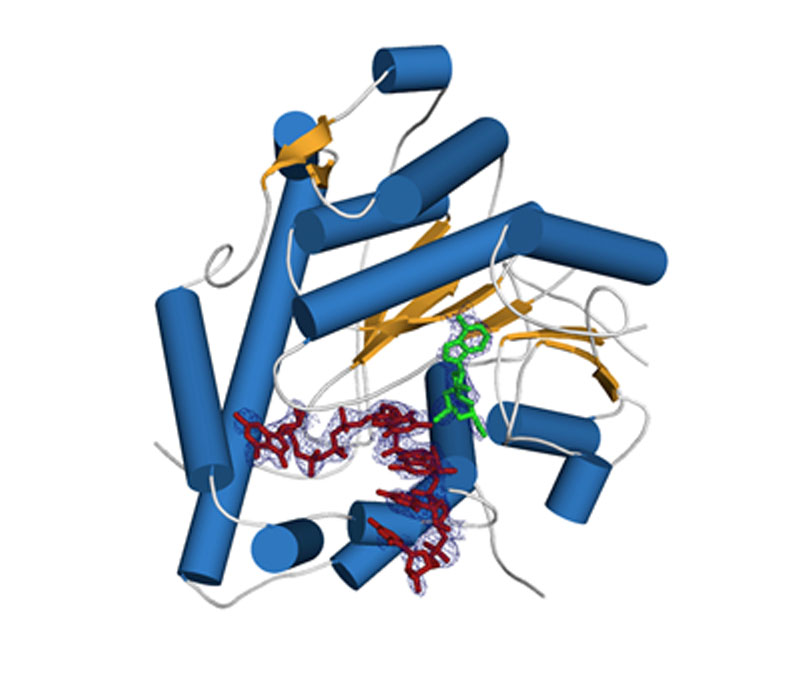
Crystal structure of human cap 2'-OH cap methyl transferase 1 (hMTR1). A fragment of capped mRNA is shown in red and the S-adenosylmethionine (methyl group donor) in green.
Mitochondrial exoribonuclease complex mtEXO
RNA degradation pathways play crucial roles in processing of various types of RNA, regulation of gene expression, and efficient removal of defective RNAs. The main executor of RNA turnover and surveillance activity in yeast mitochondria is the mtEXO complex, composed of Dss1 3ʹ-to-5ʹ exoribonuclease and Suv3 helicase.
Rażew M, Warkocki Z, Taube M, Kolondra A, Czarnocki-Cieciura M, Nowak E, Łabędzka-Dmoch K, Kawińska A, Piątkowski J, Golik P, Kozak M, Dziembowski A, Nowotny M. Structural analysis of mtEXO mitochondrial RNA degradosome reveals tight coupling of nuclease and helicase components. Nat Commun., 2018 Jan 8;9(1):97.
-
Crystal structure of Dss1 exoribonuclease from Candida glabrata reveals it is a unique member of the RNase II family with specialized domains responsible for interactions with Suv3 helicase.
-
Crystal structure of the mtEXO complex reveals the arrangement of both subunits in which the helicase motor feeds the 3' end of the RNA into the catalytic channel of Dss1 for its efficient degradation.
-
Co-operation of both helicase and nuclease activities within the complex is particularly important for degeneration of structured RNAs which cannot be handled by Dss1 on its own and for which the unwinding activity of Suc3 is required.
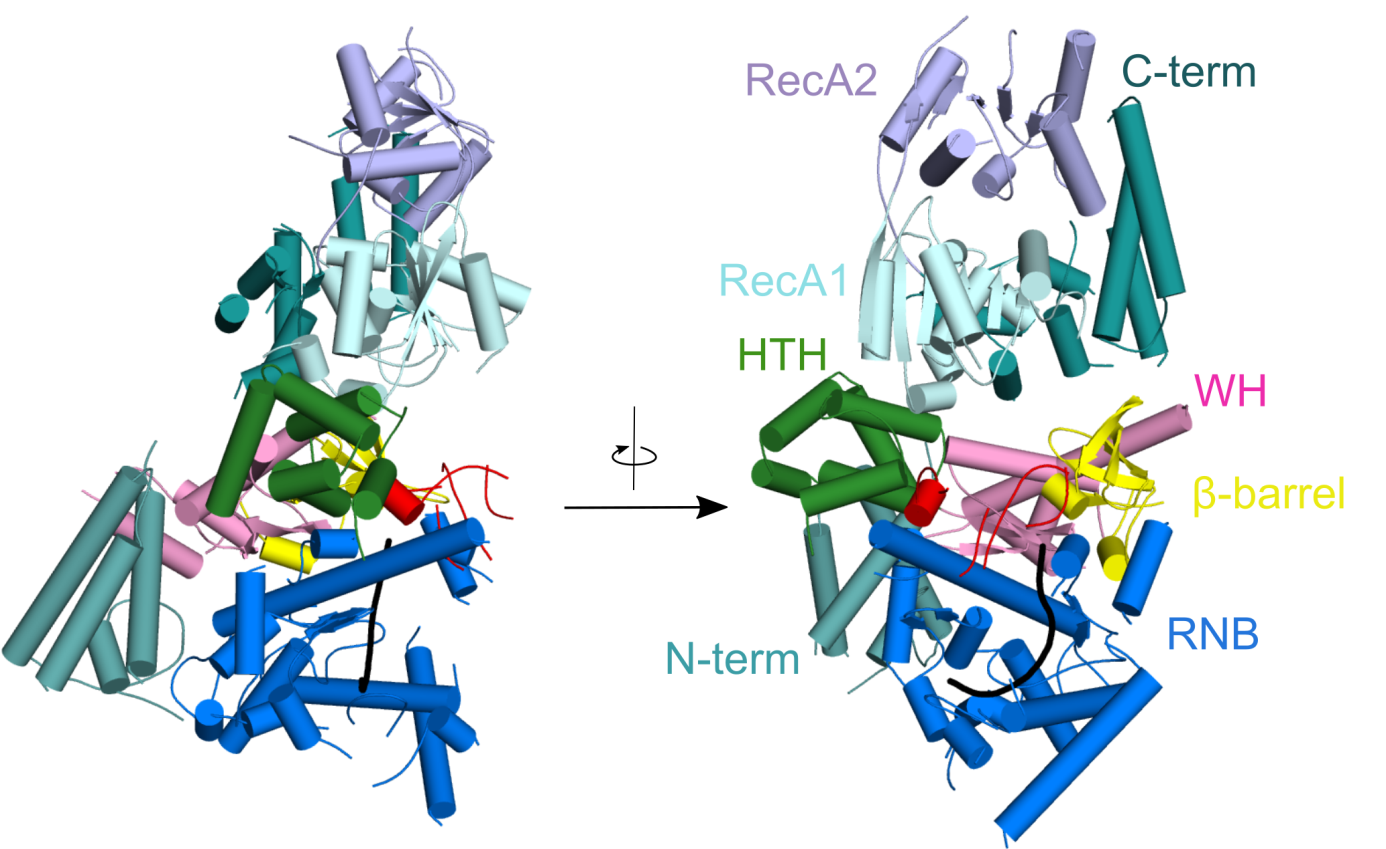
Crystal structure of Candida glabrata mtEXO complex shows the arrangement of the Suv3 helicase on top of Dss1 exoribonucleases and its accessory domains decorating the catalytic RNB domain (shown in blue) with the RNA molecule trapped inside (shown in black).
CutA terminal ribonucleotide transferase
Template-independent terminal ribonucleotide transferases (TENTs) catalyze the addition of nucleotide monophosphates to the 3′-end of RNA molecules regulating their fate. A subgroup TENTs are 3′ CUCU-tagging enzymes, such as CutA in Aspergillus nidulans. CutA preferentially incorporates cytosines, processively polymerizes only adenosines and does not incorporate or extend guanosines.
Malik D, Kobyłecki K, Krawczyk P, Poznański J, Jakielaszek A, Napiórkowska A, Dziembowski A, Tomecki R&, Nowotny M&, Structure and mechanism of CutA, RNA nucleotidyl transferase with an unusual preference for cytosine, Nucleic Acids Res., 2020, 48(16):9387-9405; & - corresponding authors
-
The first structural characterization of a TENT adding C/U tails - structures solved for CutA in complex with incoming CTP analog and RNA with three adenosines.
-
The binding of GTP or a primer with a terminal guanosine is predicted to lead to clashes between NH2 of the guanine and the protein, explaining why CutA is unable to use these ligands as substrates.
-
Processive adenosine incorporation likely results from tighter binding of the primer with 3′-terminal adenosine and efficient stacking between adenosine bases of the primer and incoming ATP.
-
A dynamic process of NTP recognition proposed.
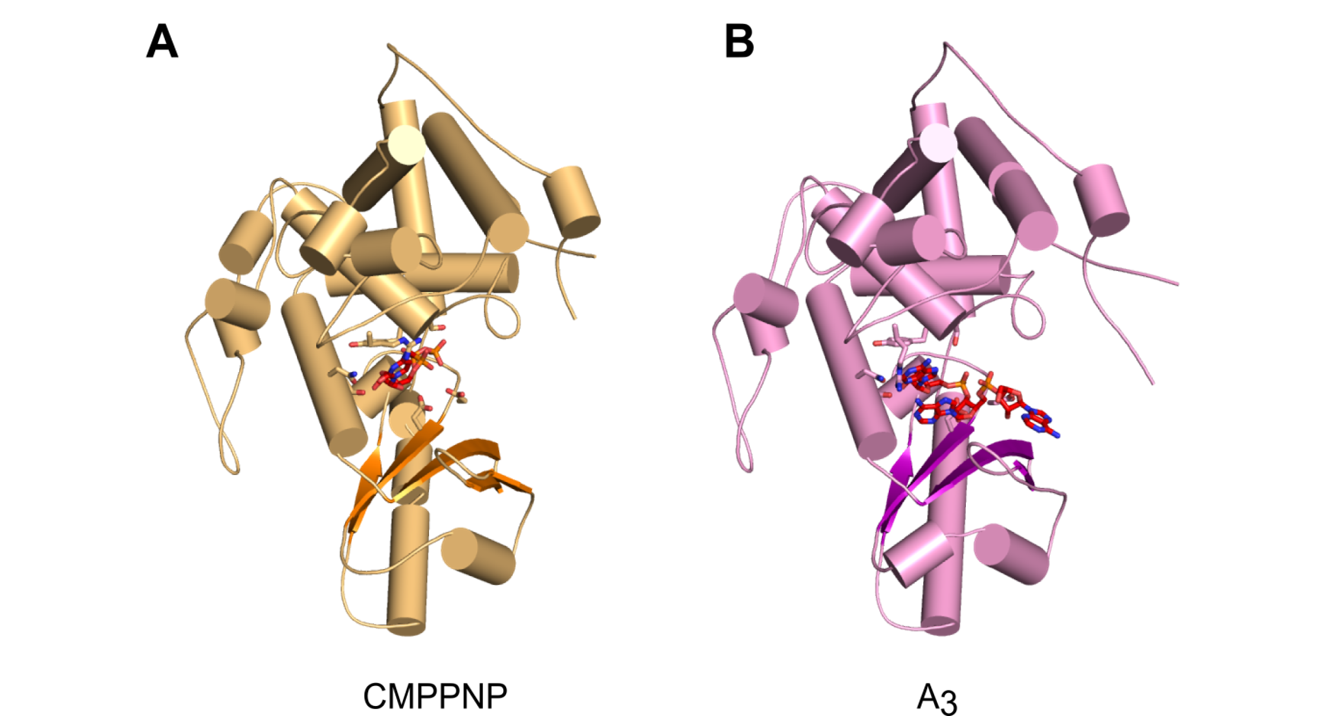
Overall structures of CutA complexes. (A) Structure of CutA–CMPCPP complex. The protein is shown as an orange cartoon, with β-strands in a darker shade of orange. Incoming nucleotide is shown as red sticks and active site residues are shown as orange sticks. (B) Structure of CutA–A3 complex. The protein is shown as pink cartoon, with β-strands in purple. The RNA is shown as red sticks and active site residues are shown as pink sticks.
5. Nucleic acid-based therapeutics
Antisense technology is an emerging therapeutic approach for inhibiting gene expression through recognition and RNase H1-dependent degradation of cellular mRNAs. In this technology, short, synthetic, single-stranded oligonucleotides (ASOs) are designed that are complementary to the target mRNA.
ASOs are frequently chemically modified for enhancing the therapeutic properties of these molecules. The most widely used modifications include phosphorothioate (PS) backbone and modifications of the sugar moieties at 2’ site. The molecular mechanism of toxicity of chemically modified antisense oligonucleotides are not fully understood.
Hyjek-Składanowska M, Vickers T, Napiórkowska A, Anderson BA, Tanowitz M, Crooke ST, Liang X, Seth PP&, Nowotny M&. Origins of the increased affinity of phosphorothioate-modified therapeutic nucleic acids for proteins. J. Am. Chem. Soc., 2020, 142 (16): 7456-7468;
& - corresponding authors
-
The first crystal structure of a complex between a protein and fully PS nucleic acid (DNA-binding domain of a model ASO-binding protein PC4 in complex with full PS 2′-OMe DNA gapmer ASO).
-
The structure reveals a possible mechanism of ASO-induced toxic protein aggregation: ASO is bound in hairpin-like conformation and its exposed gapmer part promotes the formation of a dimer of dimers of PC4 through base pairing.
-
The protein interacts with the PS-nucleic acid through a network of electrostatic and hydrophobic interactions. Importantly, the backbone of the PS ASO is able to form new and more extensive hydrophobic interactions than a natural phosphodiester backbone which provides insights into the origins for the enhanced affinity of PS for proteins.
The studies of PC4-ASO complex have been performed in cooperation with Ionis Pharmaceuticals (Carlsbad, California, USA).
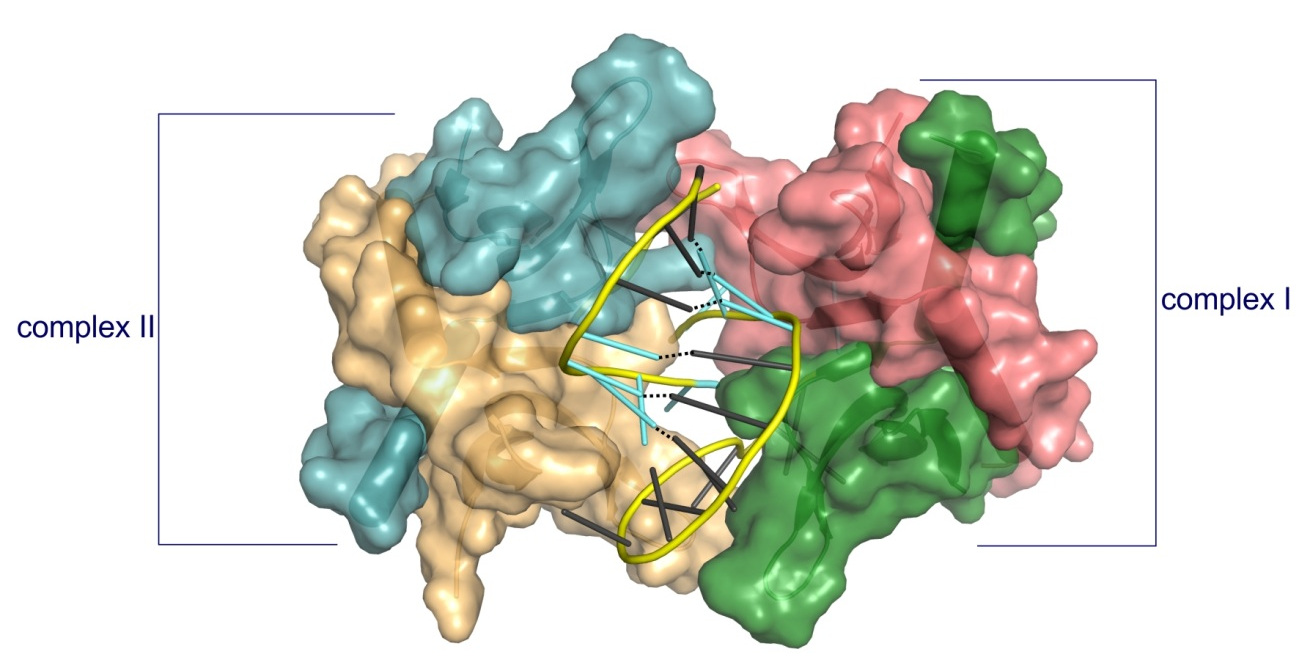
Structure of PC4 in complex with PS 2′-OMe DNA gapmer ASO. The content of the asymmetric unit of the PTEN ASO complex crystal. PC4 protomers are color-coded and shown in surface representation, DNA is shown in cartoon (PS backbone is shown in yellow, 2′-OMe PS nucleotides are shown in aquamarine, DNA gapmer nucleotides are shown in dark gray). The base pairing between the nucleotides is shown as black dotted lines.
6. Drug development projects
We cooperated with pharmaceutical companies in multiple drug development projects by performing structural studies of complexes of target proteins with inhibitors. Until the end of 2019 these projects were performed by a specialized laboratory – IIMCB Structural Biology Center with Marcin Nowotny as a CSO.
A published example of such cooperation concerns OATD-01 compound developed by OncoArendi Therapeutics company. OATD-01 is an inhibitor chitotriosidase/acidic mammalian chitinase which can be used in the treatment of idiopathic pulmonary fibrosis. The compound recently completed phase I clinical studies. We contributed the structural characterization of the complex between chitinase and the inhibitor.
Robert Koralewski, Barbara Dymek, Marzena Mazur, Piotr Sklepkiewicz, Sylwia Olejniczak, Wojciech Czestkowski, Krzysztof Matyszewski, Gleb Andryianau, Piotr Niedziejko, Michal Kowalski, Mariusz Gruza, Bartłomiej Borek, Karol Jedrzejczak, Agnieszka Bartoszewicz, Elżbieta Pluta, Aleksandra Rymaszewska, Magdalena Kania, Tomasz Rejczak, Sylwia Piasecka, Michal Mlacki, Marcin Mazurkiewicz, Michał Piotrowicz, Magdalena Salamon, Agnieszka Zagozdzon, Agnieszka Napiorkowska-Gromadzka, Aneta Bartlomiejczak, Witold Mozga, Paweł Dobrzański, Karolina Dzwonek, Jakub Golab, Marcin Nowotny, Jacek Olczak, and Adam Golebiowski, Discovery of OATD-01, a First-in-Class Chitinase Inhibitor as Potential New Therapeutics for Idiopathic Pulmonary Fibrosis. J. Med. Chem. Published October 20, 2020, https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01179#

Close-up view of active site of hCHIT1 (represented as surface colored according to electrostatic potential) in complex with OATD-01 (blue sticks).

{kind=link}