 |
Katarzyna Mleczko-Sanecka, PhD
Correspondence address:
Laboratory of Iron Homeostasis
International Institute of Molecular and Cell Biology
4 Ks. Trojdena Street, 02-109 Warsaw, Poland
Email: This email address is being protected from spambots. You need JavaScript enabled to view it.
tel: +48 (22) 597 0776; fax: +48 (22) 597 0715
|
DEGREES
2011 - PhD in Biology, European Molecular Biology Laboratory (EMBL) Heidelberg and Heidelberg University, Germany
2007 - MSc in Biotechnology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Cracow, Poland
PROFESSIONAL EXPERIENCE
2017-present - Professor, Head of Laboratory of Iron Homeostasis, International Institute of Molecular and Cell Biology in Warsaw, Poland
2011-2015 - Postdoctoral research with Prof. Martina Muckenthaler and Prof. Matthias W. Hentze, Molecular Medicine Partnership Unit, European Molecular Biology Laboratory and Heidelberg University, Heidelberg, Germany
2007-2011 - Doctoral research with Prof. Martina Muckenthaler and Prof. Matthias W. Hentze, Molecular Medicine Partnership Unit, European Molecular Biology Laboratory and Heidelberg University, Heidelberg, Germany
2006-2007 - Master thesis research with Prof. Józef Dulak and Prof. Alicja Józkowicz, Department of Medical Biotechnology, Jagiellonian University, Cracow, Poland
2006 - Undergraduate research during Erasmus fellowship with Dr. Claudine Kieda, Centre De Biophysique Moleculaire, Centre National de la Recherche Scientifique, Orleans, France
2005 - Undergraduate research during Erasmus scholarship with Dr. Claudine Kieda, Centre De Biophysique Moleculaire, Centre National de la Recherche Scientifique, Orleans, France
HONORS, PRIZES AND AWARDS
2016 - POLONEZ, National Science Centre
2014 - Independent research grant, University of Heidelberg
2011 - Invitation to 61st Lindau Meeting of Nobel Laureates, Lindau, Germany
2015, 2014, 2011-2009 - Travel Grant to attend and present data at the international conferences in iron biology
2007 - Louis-Jeantet PhD Scholarship for young researchers from Eastern Europe to support PhD studies at European Molecular Biology Laboratory
2006 - Erasmus Scholarship, Centre National de la Recherche Scientifique, Orleans, France
DESCRIPTION OF CURRENT RESEARCH
Sufficient iron supplies are critical for vital cellular functions, such as energy production and RNA/ DNA processing and repair. In the human body, the vast majority of iron is utilized for hemoglobin synthesis during the daily production of ~200 billion erythrocytes. However, an excess of free iron can cause oxidative damage and lead to organ failure. The maintenance of iron balance is thus essential for the proper functioning of cells and organisms. Broadening our knowledge of the genetic control of iron homeostasis is important for human health. The major objective of research in the Laboratory of Iron Homeostasis is to better understand the processes that impact systemic and cellular iron levels and identify new players in iron-regulatory pathways.
At the systemic level, more than 90% of daily iron needs are met by internal iron recycling from senescent erythrocytes by splenic macrophages. The iron pool in the body is largely preserved. Because iron excretion is unregulated, iron acquisition in the intestine and its release from splenic macrophage stores must be tightly controlled. These tasks are chiefly fulfilled by the master iron-regulatory hormone hepcidin.
When iron levels in the body increase, hepcidin production is enhanced to prevent further iron absorption from the diet. To gain insights into the genetic control of iron homeostasis, we previously designed and conducted large-scale RNAi screens for novel hepcidin regulators. Follow-up work of our unbiased screens (i) revealed that SMAD7 is an important hepcidin inhibitor, (ii) linked hepcidin control to proliferative signaling, and (iii) aided in the identification of two commonly prescribed drugs, the antihypertensive drug spironolactone and antineoplastic drug imatinib, as hepcidinsuppressing agents in cultured cells and mice (Mleczko-Sanecka et al., Blood, 2010; Gut, 2014; Haematologica, 2017). Nevertheless, despite growing knowledge of the molecular control of iron homeostasis, the genetic basis for variations in body iron parameters is still not fully understood. Thus, identifying elusive factors that modify such processes as iron sensing, iron flux, and iron accumulation has high medical relevance.
When iron levels in the body increase, iron-sensing mechanisms are engaged to enhance hepcidin production and prevent further dietary iron uptake. Bone morphogenetic protein 6 (BMP6) is a cytokine that is produced by liver sinusoidal endothelial cells (LSECs) and stimulates hepcidin production in hepatocytes in response to iron challenge. Despite the critical role of BMP6 in iron sensing and the maintenance of iron balance in the body, unclear are the ways in which systemic or liver iron levels translate into alterations of Bmp6 mRNA levels in LSECs. It also remains elusive how different cell types in the liver may contribute to Bmp6 regulation. To address these issues, we first employed two immortalized cell lines that were derived from murine LSECs. We found that both LSEC models efficiently took up non-transferrin bound iron (NTBI), FeNTA, and iron citrate but not iron-bound transferrin. Iron accumulation led to oxidative stress but did not induce Bmp6 mRNA expression. Physiologically, liver hepatocytes are the first cells that acquire NTBI. We speculated that communication between LSECs and hepatocytes may contribute to the control of Bmp6 transcription. We established a system in which LSEC models or primary liver cultures that contain LSECs are co-cultured with hepatocytes (i.e., Hepa1-6 cells or primary murine hepatocytes), treated with NTBI, and then separated using FACS. Compared with LSECs that were cultured alone, LSECs that were maintained in culture together with hepatocytes induced Bmp6 mRNA expression in a more pronounced manner in response to iron supplementation (Fig. 1A, B). These data indicate that a factor that is secreted by ironloaded hepatocytes rather than iron deposition in LSECs itself serves as a signal to enhance Bmp6 expression. Our initial work showed that one candidate molecule that may perform this function was extracellular ferritin. Upon iron treatment, this protein may shuffle between hepatocytes and LSECs (Fig. 1C, D) and it stimulated Bmp6 expression in LSEC models (Fig. 1E). Our future work will utilize primary liver cell cultures and mice to improve our understanding of ferritin-dependent and possibly ferritin-independent intercellular communication that is required for iron sensing in the liver.
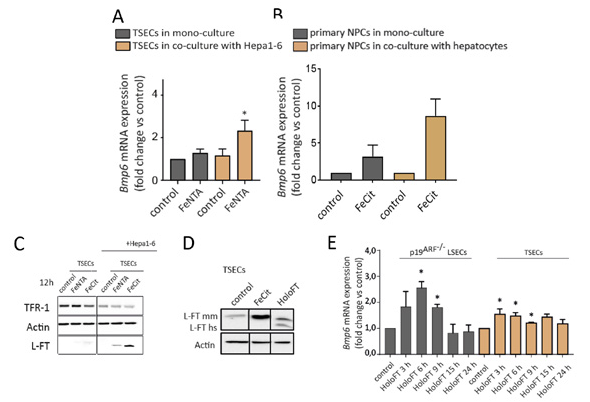
fig.1. (A) Unlike in TSECs that were cultured alone, TSECs that were maintained in culture together with hepatocytic Hepa1-6 cells induced Bmp6 mRNA expression in response to iron supplementation. (B) Liver sinusoidal endothelial cells that were sorted from primary liver cell co-cultures responded in a more pronounced manner to iron supplementation than LSECs that were cultured without hepatocytes. (C) Upon iron treatments, ferritin levels are more elevated in TSECs after co-culture with Hepa1-6 cells. (D) Human (hs) ferritin was internalized into murine TSEC cells. (E) Supporting the hypothesis that ferritin is secreted from hepatocytes to stimulate LSEC Bmp6 expression, Bmp6 mRNA levels in LSEC models were responsive to ferritin treatment.
Iron levels increase when iron challenge persists or when hepcidin responses are dysregulated, ultimately leading to the excessive saturation of transferrin and generation of NTBI. This form of “free iron” is highly toxic and currently considered the main contributor to iron-overload disorders. Liver hepatocytes are the primary cell type that acquires NTBI, which can lead to impairments in liver function and a higher risk of aggressive hepatocellular carcinoma. Hepatic iron accumulation is a hallmark of hereditary hemochromatosis and some severe anemias (e.g., thalassemias) and accompanies several other common liver diseases. Interestingly, the severity of iron loading, particularly in hemochromatosis, differs substantially between patients. The genetic basis of this variation is still not fully understood. One of our ongoing projects seeks to understand the molecular processes that contribute to NTBI uptake in hepatocytes. We are seeking to identify signaling mechanisms that control or alter the hepatic expression of ZIP14 (which is encoded by SLC39A14), the key metal transporter that is responsible for NTBI uptake in the liver. ZIP14 is considered an attractive therapeutic target to prevent or limit liver iron loading. The identification of druggable ZIP14 regulators may contribute to new pharmacological interventions and shed light on underdiagnosed iron-related side effects of some pharmaceutical compounds, which we demonstrated recently for hepcidin. Insights into regulatory mechanisms of ZIP14 may also help identify genes that modify the severity of iron overload and can serve as diagnostic markers to predict which patients are at risk of developing overt clinical symptoms. The ablation of ZIP14 in zebrafish and mice and ZIP14 mutations in humans were recently reported to lead to hepatic manganese (Mn) deficiency and Mn accumulation in other organs, notably in the brain where Mn deposition causes neurotoxicity. Thus, a comprehensive characterization of the ZIP14 regulatory network has medical relevance to our understanding of Mn homeostasis.
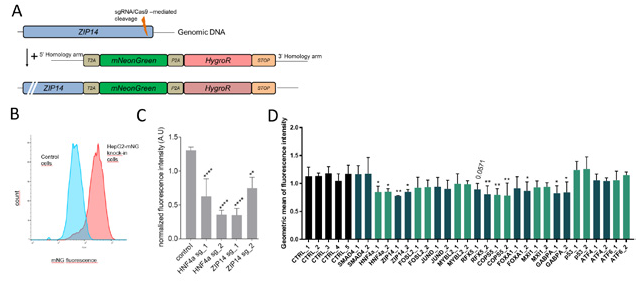
fig.2. Generation, validation, and utilization of fluorescent ZIP14 reporter HepG2 cells. (A) Shown is the ZIP14 locus with CRISPR-mediated knock-in of the fluorescent mNeonGreen (mNG) gene together with the hygromycin resistance gene. (B, C) The fluorescence signal intensity from engineered cells decreased upon CRIPSR knockout of the ZIP14 regulator HNF4a and ZIP14 itself. (D) CRISPR-mediated knockout in engineered fluorescent cells aided the identification of new ZIP14 regulators.
To decipher the ZIP14 regulome, we aim to apply state-of-the-art CRISPR-based genetic screens, followed by functional characterization of the most interesting hits in cellular assays and mice. We have already employed CRISPR-based geneediting technology to generate reporter cells that are engineered to monitor endogenous levels of ZIP14 using a fluorescence-based readout (Fig. 2A, B). We validated this new-generation reporter system by showing that CRISPR-based depletion of the newly identified ZIP14 regulator, HNF4α, as well as ZIP14 itself, efficiently reduced the fluorescent signal, reflecting the response of endogenous ZIP14 mRNA (Fig. 2C). To expand our knowledge of the transcriptional control of ZIP14 and possibly identify additional genes that can serve as positive controls for our future screens, we analyzed the promoter region of the ZIP14 gene using available chromatic accessibility and ChIPseq data. This approach allowed us to identify a region that is further upstream of the transcription start site where liver-enriched transcription factors bind. Together with hints from transcriptomics databases, this guided us to a priori select several potential genes that may be involved in the control of ZIP14 mRNA expression levels. These genes were further verified as ZIP14 regulators using our engineered fluorescent cell line, thus validating three of the genes (COPS5, RFX5, and GABPA; Fig. 2D). Our further work will help us to better understand the involvement of these and other genes in ZIP14 regulation.

Lab Leader:
PhD Students:
Lab Technician:
Laboratory Support Specialist: